01. Defects in Sr2FeMoO6.
R. Mishra, O. D. Restrepo, P. M. Woodward, W. Windl, Chem. Mater. 22, 6092 (2010).
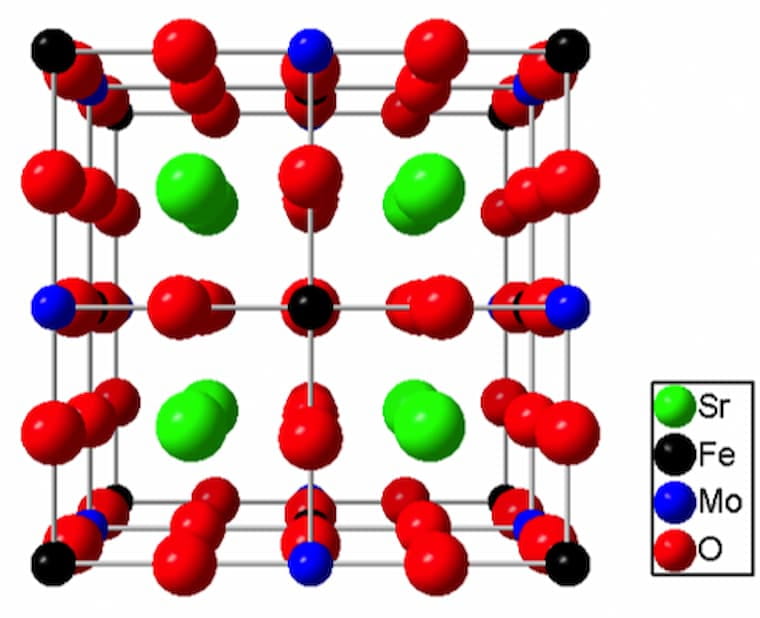
First principles study of defective and non-stoichiometric Sr2FeMoO6.
R. Mishra, O. D. Restrepo, P. M. Woodward, W. Windl, Chem. Mater. 22, 6092 (2010).
The influence of disorder and stoichiometry-breaking point defects on the structural and magnetic properties of Sr2FeMoO6 have been investigated with the help of electronic structure calculations within the spin-polarized GGA+U approach. Defining the chemical potentials of the constituent elements from constitutional defects, we calculate the energetics of the possible point defects in nonstoichiometric Sr2FeMoO6 and find transition-metal-ion antisites and oxygen vacancies to be dominant. In nonstoichiometric Sr2Fe1-xMo1-xO6 with -0.75 < x < 0.25, both FeMo antisites (for Fe-rich samples or x > 0) and MoFe antisites (for Mo-rich samples or x < 0) lead to a systematic decrease in saturation magnetization. Only MoFe antisites destroy the half-metallic character of the electronic structure, since their t2g band crosses the Fermi level for x e -0.125. This leads to a decrease of spin polarization from 100% for x > -0.125 to 0 at x ≈ -0.75. Oxygen vacancies also reduce the saturation magnetization, but the half-metallic character and, hence, 100% spin polarization is retained. The optimized unit-cell lattice parameter remains within a relatively narrow range (7.96 A˚ for x = 0.25 to 8.00 A˚ for x = -0.75), despite large changes in composition. In stoichiometric Sr2FeMoO6, the saturation magnetization decreases linearly as the Fe/Mo antisite disorder increases, and the half metallicity is lost, because of the t2g states on both MoFe and FeMo. The spin polarization remains ∼100% only for very small amounts of disorder. The calculated disorder formation energies suggest that short-range ordering is favorable in Sr2FeMoO6. The calculated results are in excelle