11. Tunable gaps and enhanced mobilities in silicane
O. D. Restrepo, R. Mishra, J. E. Goldeberger, W. Windl, J. Appl. Phys. 115, 033711 (2014).
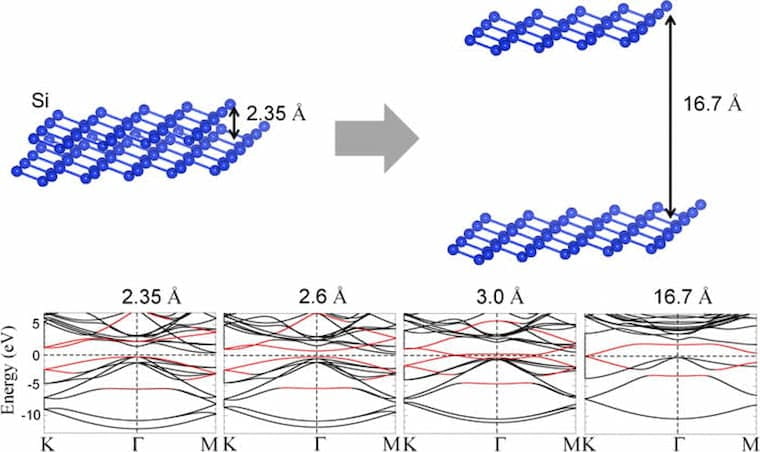
Tunable gaps and enhanced mobilities in strain-engineered silicane.
O. D. Restrepo, R. Mishra, J. E. Goldeberger, W. Windl, J. Appl. Phys. 115, 033711 (2014).
The recent demonstration of single-atom thick, sp3-hybridized group 14 analogues of graphene enables the creation of materials with electronic structures that are manipulated by the nature of the covalently bound substituents above and below the sheet. These analogues can be electronically derived from isolated (111) layers of the bulk diamond lattice. Here, we perform systematic Density Functional Theory calculations to understand how the band dispersions, effective masses, and band gaps change as the bulk silicon (111) layers are continuously separated from each other until they are electronically isolated, and then passivated with hydrogen. High-level calculations based on HSE06 hybrid functionals were performed on each endpoint to compare directly with experimental values. We find that the change in the electronic structure due to variations in the Si-H bond length, Si-Si-Si bond angle, and most significantly the Si-Si bond length can tune the nature of the band gap from indirect to direct with dramatic effects on the transport properties. First-principles calculations of the phonon-limited electron mobility predict a value of 464 cm2/Vs for relaxed indirect band gap Si-H monolayers at room temperature. However, for 1.6% tensile strain, the band gap becomes direct, which increases the mobility significantly (8 551 cm2/Vs at 4% tensile strain). In total, this analysis of Si-based monolayers suggests that strain can change the nature of the band gap from indirect to direct and increase the electron mobilitymore than 18-fold.